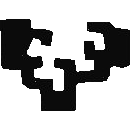Working with proteomics data in R
Proteomics in Biomedicine
September 29, 2025
First of all …
- Click the following link to start downloading a file (15 minutes) we will need:
- Already downloaded at
C:\Usuarios\Acceso Publico\Documentos Publicos
1 Goal
- Understand the steps and data structures of a shotgun proteomics bioinformatics workflow:
- After the search engine (already covered in previous sessions)
- In a practical way with a computer
- Using free/open-source solutions instead of “black box” software:
- R notebooks
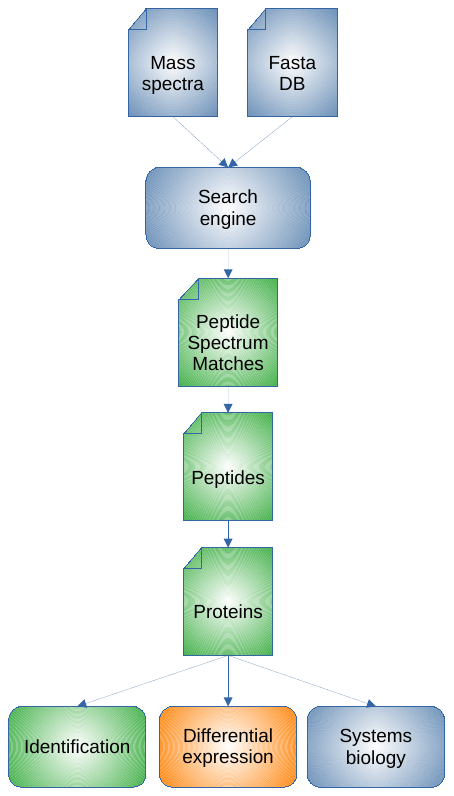
2.4 Tidyverse
- Will work with data (PSMs, peptides, proteins, etc.) organized in tibbles (≈dataframes):
- Like a spreadsheet, with rows (observations) and columns (variables)
- Tidyverse provides useful packages for data science:
- See R for Data Science for a great book (free)
3.1 Virtual machine
- Launch VirtualBox and import (~2 minutes) the downloaded file (
*.ova)
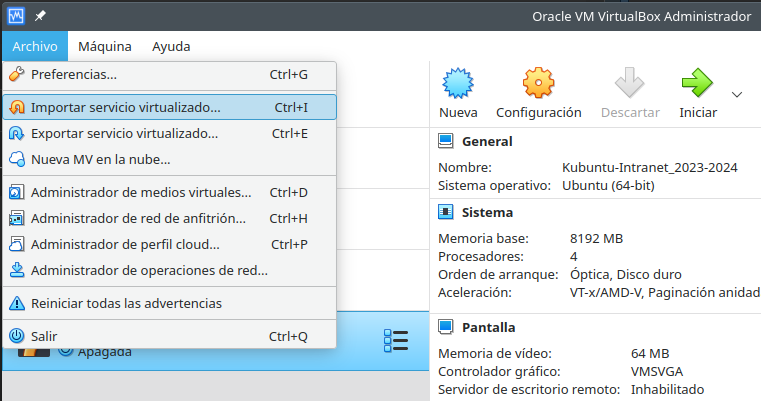
Configure the virtual machine hardware according to your available resources
Start the virtual machine and log-in using your LDAP credentials
3.2 R Studio
- Click on Projects/R/ProteomicsBiomedicine/R.Rproj to open the R Studio project provided
- Now you can open the latest version of the provided notebooks:
introduction.Rmd(this document)workflow_id.Rmd:
- Or even create a new notebook for practicing:
- New file/R Notebook